Jean-Louis SCHLIENGER, Université de Strasbourg
Découvert en 1923, il y a un siècle, alors qu’il n’était pas recherché, dans le sillage des travaux effectués sur l’insuline qui venait d’être isolée en 1921 par Banting et Best au laboratoire de MacLeod à l’Université de Toronto, le glucagon a connu un destin en demi-teinte. Contrairement à son hormone jumelle, l’insuline, le « principe hyperglycémiant » n’a pas bouleversé le traitement d’une maladie. Son rôle physiologique, longtemps cantonné à la production hépatique de glucose, a été sous-estimé tout comme son potentiel thérapeutique. La transition d’un statut initial d’impureté polluant la préparation de l’insuline à celui de possible auxiliaire thérapeutique dans le diabète a été chaotique. Le glucagon que l’on croyait voué à la seule correction des hypoglycémies sévères connaît à présent un regain d’intérêt dans le traitement du diabète et de l’obésité à la suite du développement d’agonistes de son récepteur
Découvert en deux temps, le glucagon est un fruit de la sérendipité
La première manifestation du glucagon a été accidentelle et totalement incomprise. Banting et Best qui s’acharnaient à isoler le principe hypoglycémiant du pancréas durant le torride été 1921 avaient observé une réponse biphasique de la glycémie lors de l’injection de broyats de pancréas à des chiens pancréatectomisés, l’hypoglycémie souhaitée étant précédée d’une discrète hyperglycémie transitoire qu’ils prirent le parti d’ignorer en l’attribuant à un artefact.
Comme d’autres, Charles P. Kimball et John Raymond Murlin, de l’Université de Rochester (États-Unis) s’attachèrent à optimiser la production d’insuline devenue un médicament vedette. Ils mirent au point une méthode peu coûteuse d’extraction et de concentration de l’insuline quelques mois après le groupe de Toronto, mais à la différence de Banting, Murlin (1874-1960) prêta attention à l’hyperglycémie transitoire survenant immédiatement après l’injection de l’insuline (figure 1). En octobre 1922, il parvint à séparer une fraction hyperglycémiante soluble dans l’alcool de la fraction hypoglycémiante insoluble riche en insuline. Son injection sous-cutanée à un chien provoquait une élévation de la glycémie de 50 à 100 % pendant 3 heures. Murlin et ses collaborateurs démontrèrent que l’hyperglycémie était la conséquence d’un principe actif hyperglycémiant présent dans le pancréas en même temps que l’insuline et le dénommèrent « glucagon » par contraction des termes « glucose » et « agoniste », sans poursuivre davantage leurs travaux(1). Ce n’est qu’en 1935 qu’une équipe de l’Université de Bonn identifia dans le pancréas une substance « extrêmement similaire à l’insuline par ses propriétés chimiques et physiques… ayant une structure protéique (et) une teneur en C, H, N et S comparable à celle de l’insuline » ayant une action hyperglycémiante franche attribuée à un effet glycogénolytique hépatique baptisé « facteur HG », le nom de glucagon étant passé à la trappe(2).
Découvert en 1923, il y a un siècle, alors qu’il n’était pas recherché, dans le sillage des travaux effectués sur l’insuline qui venait d’être isolée en 1921 par Banting et Best au laboratoire de MacLeod à l’Université de Toronto, le glucagon a connu un destin en demi-teinte. Contrairement à son hormone jumelle, l’insuline, le « principe hyperglycémiant » n’a pas bouleversé le traitement d’une maladie. Son rôle physiologique, longtemps cantonné à la production hépatique de glucose, a été sous-estimé tout comme son potentiel thérapeutique. La transition d’un statut initial d’impureté polluant la préparation de l’insuline à celui de possible auxiliaire thérapeutique dans le diabète a été chaotique. Le glucagon que l’on croyait voué à la seule correction des hypoglycémies sévères connaît à présent un regain d’intérêt dans le traitement du diabète et de l’obésité à la suite du développement d’agonistes de son récepteu
Découvert en deux temps, le glucagon est un fruit de la sérendipité
La première manifestation du glucagon a été accidentelle et totalement incomprise. Banting et Best qui s’acharnaient à isoler le principe hypoglycémiant du pancréas durant le torride été 1921 avaient observé une réponse biphasique de la glycémie lors de l’injection de broyats de pancréas à des chiens pancréatectomisés, l’hypoglycémie souhaitée étant précédée d’une discrète hyperglycémie transitoire qu’ils prirent le parti d’ignorer en l’attribuant à un artefact.
Comme d’autres, Charles P. Kimball et John Raymond Murlin, de l’Université de Rochester (États-Unis) s’attachèrent à optimiser la production d’insuline devenue un médicament vedette. Ils mirent au point une méthode peu coûteuse d’extraction et de concentration de l’insuline quelques mois après le groupe de Toronto, mais à la différence de Banting, Murlin (1874-1960) prêta attention à l’hyperglycémie transitoire survenant immédiatement après l’injection de l’insuline (figure 1). En octobre 1922, il parvint à séparer une fraction hyperglycémiante soluble dans l’alcool de la fraction hypoglycémiante insoluble riche en insuline. Son injection sous-cutanée à un chien provoquait une élévation de la glycémie de 50 à 100 % pendant 3 heures. Murlin et ses collaborateurs démontrèrent que l’hyperglycémie était la conséquence d’un principe actif hyperglycémiant présent dans le pancréas en même temps que l’insuline et le dénommèrent « glucagon » par contraction des termes « glucose » et « agoniste », sans poursuivre davantage leurs travaux(1). Ce n’est qu’en 1935 qu’une équipe de l’Université de Bonn identifia dans le pancréas une substance « extrêmement similaire à l’insuline par ses propriétés chimiques et physiques… ayant une structure protéique (et) une teneur en C, H, N et S comparable à celle de l’insuline » ayant une action hyperglycémiante franche attribuée à un effet glycogénolytique hépatique baptisé « facteur HG », le nom de glucagon étant passé à la trappe(2).

Figure 1. John Raymond Murlin (1874-1960) de l’Université de Rochester, qui a donné son nom au glucagon en 1923. Mais le vicomte Christian de Duve (1917-2013) de l’Université de Louvain peut être considéré comme le second « père » du glucagon par l’importance de ses travaux qui ont remis en selle le glucagon quelque peu oublié pendant un quart de siècle.
En 1948, utilisant une nouvelle méthode de cristallisation, deux futurs prix Nobel Earl W. Sutherland (1915-1974) et Christian de Duve (1917-2013) qui travaillaient de conserve à Saint-Louis (Missouri) parvinrent à localiser le facteur HG, contaminant des insulines commerciales, dans les cellules des îlots de Langerhans (ce qui sera confirmé en 1962 par des techniques d’immunofluorescence) et conclurent que le facteur HG n’était autre que le glucagon de Murlin (figure 2)(3). D’emblée, de Duve émit l’hypothèse que l’homéostasie glucosée dépendait d’une balance entre l’insuline et le glucagon, l’hypoglycémie induite par l’insuline stimulant la sécrétion de glucagon pour mobiliser les réserves glycogéniques. Un quart de siècle après Murlin, Christian de Duve (prix Nobel de physiologie ou médecine en 1974 pour ses travaux sur le lysosome et le peroxysome) donna au glucagon son statut d’hormone à la suite de travaux complémentaires menés à l’Université de Louvain. À ce titre, il peut être considéré comme le deuxième père du glucagon. Au cours des années 1950, la cristallisation, le séquençage et la synthèse du glucagon ouvrent une ère moderne de l’exploration du glucagon aboutissant à la mise au point d’un dosage radioimmunologique par Roger Unger (1924-2020) qui sera déterminant pour la compréhension du rôle physiopathologique du glucagon(4).
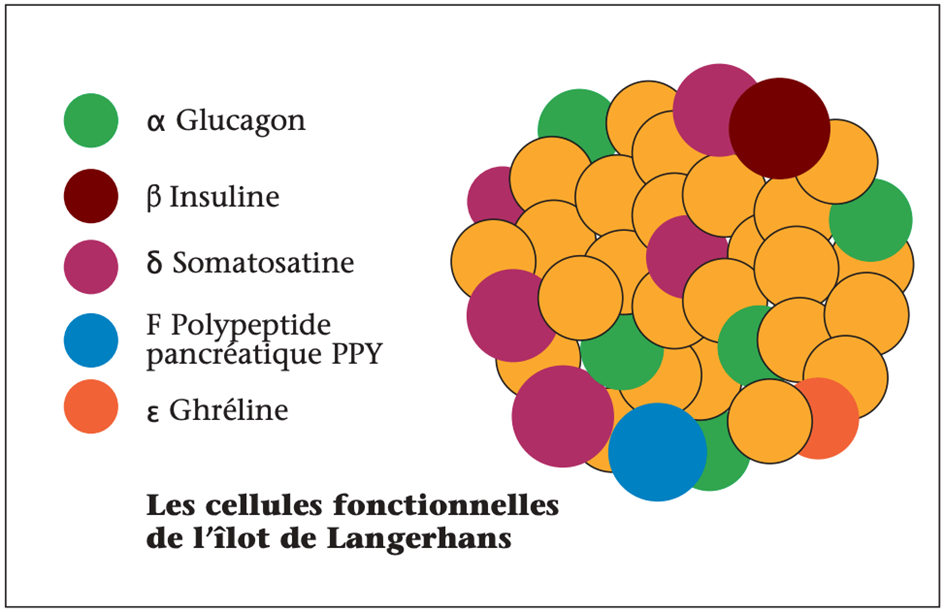
Figure 2. Cellules endocrines contenues dans l’îlot de Langerhans qui interagissent entre elles selon un mode paracrine.
Rôle physiologique du glucagon
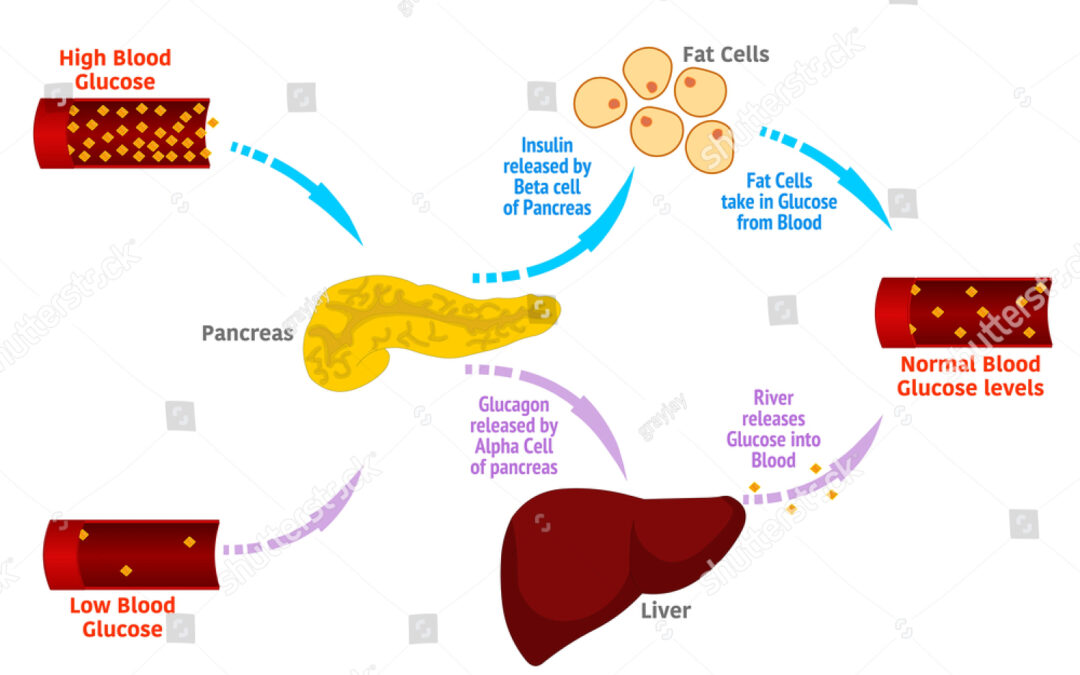
L’exploration du mécanisme d’action du glucagon et de son rôle physiopathologique bat son plein au cours des années 1960. C’est à partir de ses travaux sur le glucagon qu’Earl Sutherland a découvert le deuxième messager AMP cyclique qui lui vaudra le prix Nobel en 1971. Le glucagon fait partie des peptides issus du proglucagon, un précurseur de 160 acides aminés exprimé dans les cellules alpha pancréatiques, les cellules L entéro-endocrines et, dans une moindre mesure, dans l’hypothalamus et le tronc cérébral. Dans les cellules α, une convertase tissu spécifique aboutit à la formation de glucagon (figure 3). Grâce à un dosage RIA, Roger Unger, chercheur anticonformiste et visionnaire, a décrit les variations de la glucagonémie dans diverses situations, notamment au cours du diabète. La concentration plasmatique de glucagon inférieure à 200 pg/mL à l’état basal augmente jusqu’à cinq fois lors d’une hypoglycémie induite par l’insuline et de plus de 10 fois après une perfusion d’arginine. Les travaux expérimentaux et cliniques ont positionné le glucagon comme un agent de maintien à un niveau convenable du fuel énergétique majeur qu’est le glucose dans diverses circonstances telles que l’exercice physique, le jeûne et l’hypoglycémie(5).
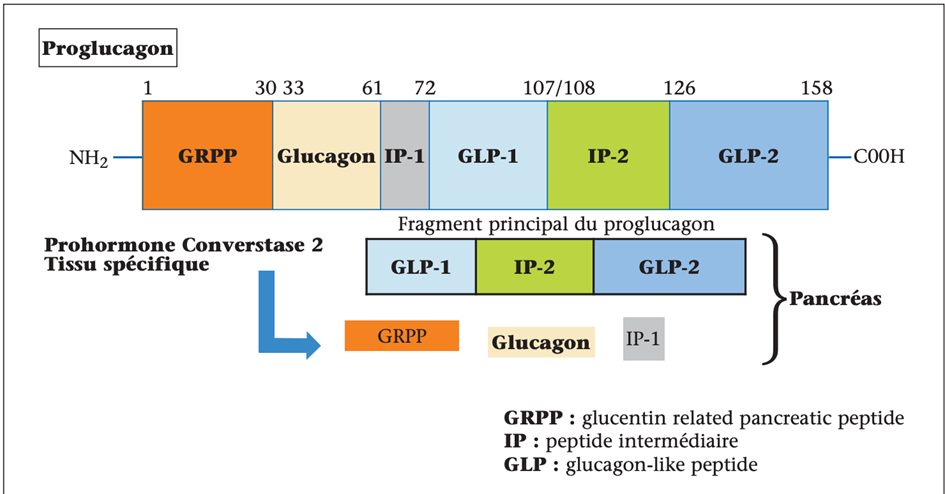
Figure 3. Conversion du proglucagon en glucagon dans les cellules α des îlots pancréatiques sous l’effet d’une convertase tissu-spécifique.
La sécrétion du glucagon est modulée par de nombreux facteurs (somatostatine sécrétée par les cellules δ, GABA, amyline, zinc, signaux du système nerveux central, acides aminés comme l’arginine, acides gras libres, etc.), mais est dominée par un feedback entre l’insuline et le glucagon, l’insuline inhibant le glucagon et le glucagon stimulant l’insuline. La diminution de la glycémie en dessous des valeurs normales est le principal déclencheur de sa sécrétion. Le rôle des incrétines mérite une mention particulière : le GLP-1 déprime la sécrétion des cellules α par un effet indirect sur les cellulesβ alors que le GIP stimule le glucagon en cas d’hypoglycémie et l’inhibe en cas de normo ou d’hyperglycémie (figure 4).
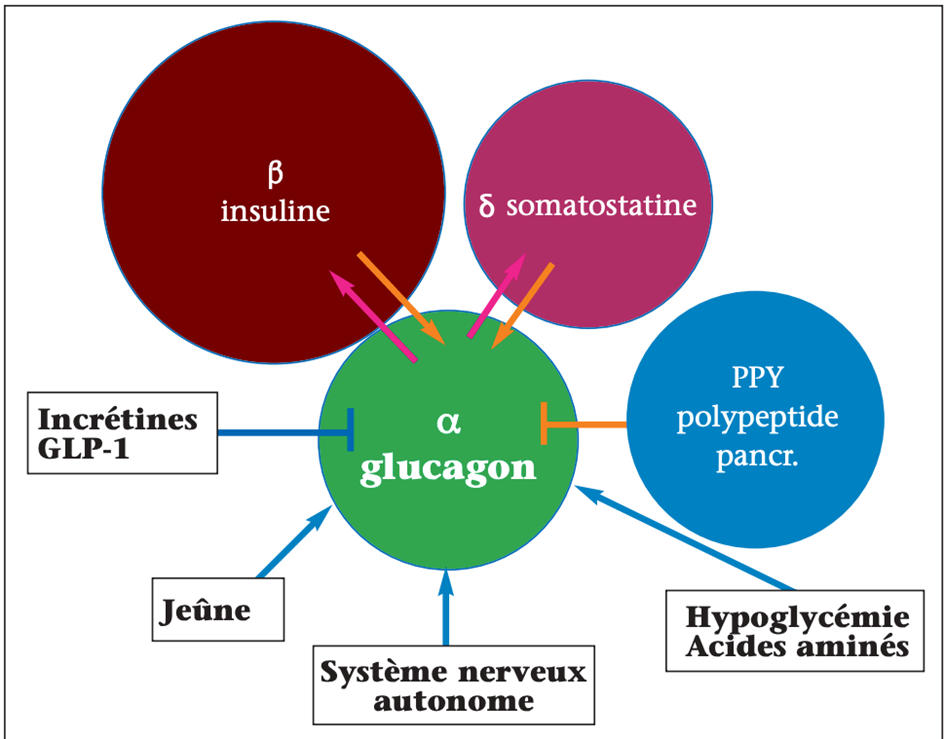
Figure 4. Régulation paracrine, autocrine et environnementale de la sécrétion du glucagon.
L’étude des mécanismes d’action du glucagon a été à l’origine de la découverte des récepteurs membranaires et du « second messager » qui assure la transduction du signal hormonal en initiant les réactions intracellulaires. En 1983, le clonage du gène codant pour le proglucagon a contribué à élucider les différents processus posttranslationnels dans les îlots pancréatiques et l’intestin. Le glucagon agit en se liant à un récepteur spécifique appartenant à la famille des récepteurs couplés aux protéines G, caractérisé en 1993, présent sur les cellulesβ pancréatiques, au niveau du foie, du tissu adipeux, des muscles lisses intestinaux et des reins.
Les effets physiologiques du glucagon s’exercent principalement dans le foie où il favorise la production de glucose par une action glycogénolytique et dans le tissu adipeux où il a un effet lipolytique à des doses pharmacologiques. En dehors de son effet sur la régulation du glucose, le glucagon exerce d’autres actions liées au métabolisme. Ses effets sur l’oxydation hépatique des acides gras et la lipogenèse intrahépatique de novo sont clairement démontrés. Il joue un rôle important dans la régulation du métabolisme des acides aminés qui eux-mêmes stimulent la sécrétion de glucagon.
Il possède de surcroît un effet thermogénique et a la propriété de stimuler la dépense énergétique dans le tissu adipeux brun. Le glucagon est aussi l’hormone du jeûne qui s’oppose aux effets de l’insuline. La diminution du rapport insuline/glucagon induite par la privation alimentaire ou en glucose déclenche une cascade métabolique adaptative comportant l’activation successive de la glycogénolyse, de la lipolyse, de la néoglucogenèse et de la cétogenèse(6). Par ailleurs, il existe un lien entre le cerveau et le glucagon qui semble médié par l’hypoglycémie et l’activation des neurones gluco-sensibles connectés aux cellules α par l’intermédiaire du système vagal ce qui détermine une diminution de la prise alimentaire en cours de repas. Dans le rein, le glucagon participe au transport transépithélial des solutés, mais ne semble pas intervenir directement sur la néoglucogenèse rénale. De plus, il possède une action inotrope et chronotrope positive utilisée en thérapeutique à des doses pharmacologiques pour restaurer la fonction cardiaque après un surdosage en β-bloquants.
Au total, le glucagon joue un rôle crucial dans la régulation glucosée, l’adaptation au jeûne et à la dénutrition et à l’exercice physique comme partenaire d’un système bi-hormonal où l’insuline favorise le stockage du glucose dans le muscle et le foie et où le glucagon augmente la production de glucose hépatique pour assurer un apport glucosé satisfaisant au cerveau en toutes circonstances.
Glucagon et pathologie
Glucagonome
Le glucagonome, seule affection pathologique directement imputable au glucagon, est une tumeur neuroendocrine développée à partir des cellules α des îlots pancréatiques responsables d’une hypersécrétion non régulée de glucagon. Le tableau clinique typique associe un érythème nécrolytique migrateur (éruption cutanée rouge, bulleuse et migratoire, associée à un prurit intense, principalement localisée aux mem bres inférieurs et à l’aine), un diabète sucré habituellement insulinorequérant, une anémie, une perte de poids, des lésions muqueuses (glossite, chéilite, stomatite), un sur-risque thromboembolique, des symptômes gastro-intestinaux (douleurs abdominales, diarrhée) et neuropsychiatriques (dépression). Son incidence est de 1/20 000 000, loin derrière celle des autres tumeurs endocrines pancréatiques. Il peut être associé à une néoplasie endocrinienne multiple de type 1 (NEM1). Des métastases hépatiques sont habituellement présentes au moment du diagnostic. Les analogues de la somatostatine (octréotide ou lanréotide) sont généralement efficaces pour améliorer les symptômes et entraîner une rémission des éruptions cutanées. La réduction chirurgicale suivie d’une chimiothérapie est indiquée dans les tumeurs étendues. Le pronostic du glucagonome est médiocre(7).
Diabète
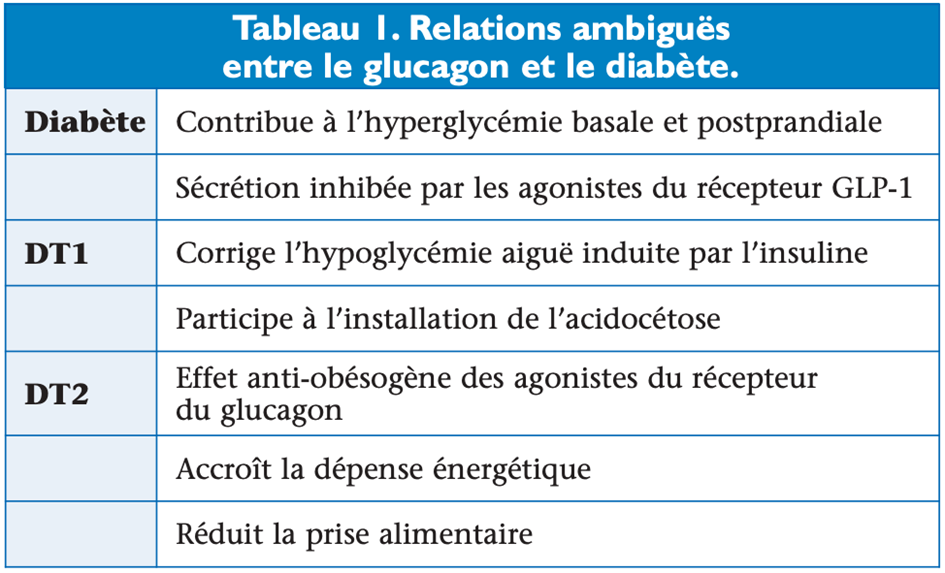
L’hyperglucagonémie absolue ou relative est un trait du diabète commun aux types 1 et 2. Son rôle pathogénique reste discuté. Dans le DT2, l’hyperglucagonémie présente à l’état basal augmente paradoxalement en postprandial et persiste en dépit de l’hyperglycémie. Responsable d’une production hépatique de glucose inappropriée, elle est liée pour une part au déficit de sécrétion de l’insuline et de somatostatine dans les îlots de Langerhans. L’administration de somatostatine, un inhibiteur de la sécrétion de glucagon produit par les cellules δ dans les îlots de Langerhans, réduit l’hyperglycémie du DT2 et l’inactivation des récepteurs du glucagon, prévient le diabète expérimental induit par la streptozotocine. Ces données expérimentales suggèrent que le diabète est une maladie bi-hormonale, l’hyperglucagonémie étant indispensable à l’expression des troubles métaboliques du diabète(8). Dans le DT1, l’hyperglucagonémie est réduite, mais ne disparaît pas lorsqu’une insulinothérapie rétablit la normoglycémie(9). Dans le DT2, elle est attribuée à une carence en insuline, à une résistance des cellules α à l’insuline ou à une moindre réactivité à l’hyperglycémie du fait d’une désensibilisation secondaire à la glucotoxicité. Dans le diabète pancréatoprive, l’hyperglucagonémie est la conséquence d’une hyperproduction de glucagon par les cellules α du tractus intestinal (tableau 1).
Hypoglycémie
Le glucagon est un élément essentiel de la contre-régulation glycémique. À la phase précoce de l’hypoglycémie induite par l’insuline, son taux augmente rapidement d’un facteur 2 à 3. Il en résulte une glycogénolyse, précédant l’activation de la néoglucogenèse, qui peut suffire à corriger l’hypoglycémie. L’hypoglycémie augmente la sensibilité du foie au glucagon (figure 5).
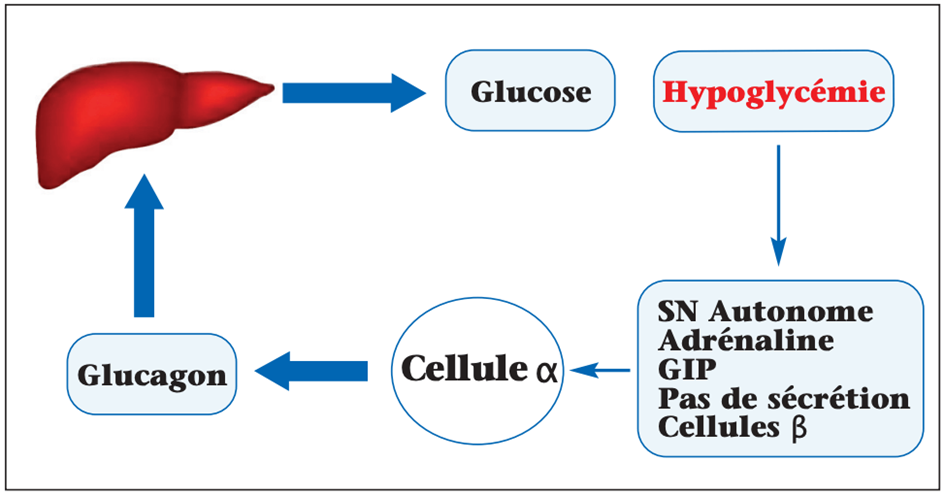
Figure 5. La stimulation de la glycogénolyse par le glucagon permet de corriger l’hypoglycémie insulinique aiguë.
Obésité
Il n’existe pas d’anomalie bien caractérisée de la régulation du glucagon dans l’obésité commune. Expérimentalement, le glucagon a la propriété d’augmenter la dépense énergétique et de diminuer les apports alimentaires. L’hyperglucagonémie n’est présente qu’en cas d’insulinorésistance marquée et de perturbation du métabolisme glucosé(10).
Glucagon et thérapeutique
Traitement des hypoglycémies sévères
En 1957, le glucagon a d’abord été utilisé en milieu psychiatrique pour interrompre le coma hypoglycémique lors de la cure de Sackel qui réalisait un choc hypoglycémique destiné à traiter la schizophrénie avant l’ère des neuroleptiques. Le procédé fut aussitôt adopté pour traiter les hypoglycémies sévères survenant chez les sujets DT1 sous insuline. À ce jour, la correction des hypoglycémies sévères reste l’indication thérapeutique majeure du glucagon administré par voie injectable ou par voie nasale.
Maladies métaboliques
Le développement d’analogues du glucagon à action prolongée, d’agonistes doubles et triples unimoléculaires chimériques et de nouvelles stratégies de ciblage dans les tissus exprimant les récepteurs du glucagon ont ouvert de nouveaux horizons thérapeutiques pour une hormone utilisée pendant des lustres pour son potentiel hyperglycémiant « aigu ».
Diabète
L’inactivation du récepteur du glucagon par des anticorps monoclonaux ou des antinucléotides « antisens » a été une piste explorée en un premier temps pour réduire la glycémie en réduisant la glycogénolyse. Les knock-out, les knock-down ou l’inhibition pharmacologique du récepteur du glucagon se sont effectivement avérés efficaces dans les modèles animaux. Des études cliniques dans le DT2 ont confirmé l’efficacité des antagonistes à base de petites molécules et de peptides pour diminuer la glycémie et réduire les excursions glycémiques sans augmenter le risque d’hypoglycémie. En dépit d’une tolérance assez satisfaisante, le développement clinique des premiers peptides candidats n’a pas été poursuivi en raison de la survenue d’une hyperplasie des cellules α et de lésions hépatiques dans les modèles murins(11). La voie des agonistes s’est avérée paradoxalement plus prometteuse. Les agonistes des récepteurs du glucagon-like peptide-1 (AR-GLP-1), qui stimulent la sécrétion de l’insuline et freinent celle du glucagon de façon glycémie-dépendante ont été la première étape. L’effet incrétine (gliptines et AR-GLP-1) a l’avantage de stimuler la sécrétion d’insuline tout en inhibant celle du glucagon, ce qui ne peut qu’être favorable dans la gestion de l’hyperglycémie. Le développement de co-agonistes visant le récepteur du GLP-1 et le récepteur du glucagon est une autre approche intéressante puisqu’elle est censée combiner les effets antidia – bétiques et anti-obésité. La coadministration d’agonistes des récepteurs du GLP-1 et du glucagon permet de contrebalancer l’hyperglycémie induite par le glucagon par les effets insulinotropes glucose-dépendants du GLP-1 tout en préservant les effets positifs du glucagon sur la dépense énergétique. Chez l’homme la co-administration de ces agonistes réduit de manière synergique l’apport alimentaire et permet de réduire la dose de GLP-1(12). Puisque le co-agonisme des récepteurs du GLP-1 et du glucagon élargit la fonctionnalité thérapeutique, l’idée est venue de développer des doubles ou triples agonistes chimériques équilibrés avec un seul profil pharmacocinétique. Ces co-agonistes paraissent également indiqués dans la stéatose hépatique non alcoolique. Des triples agonistes glucagon, GIP et GLP-1, associant les effets des incrétines au glucagon, sont d’ores et déjà développés, comme le rétatrutide LY3437943(13), dans le but d’une prise en charge plus physiologique du DT2 en assurant une perte de poids remarquable(14).
Obésité
Les analogues du glucagon à action prolongée ont des effets confirmés expérimentalement sur la réduction du poids, des ingestas et de la cholestérolémie associés à une augmentation de la dépense énergétique et de la production hépatique de FGF21 qui est un facteur de croissance apparenté à une hormone qui stimule la dépense énergétique dans le tissu adipeux brun, améliore la sensibilité à l’insuline et prévient la formation de la stéatose hépatique(15). L’augmentation de la dépense énergétique semble liée à l’augmentation du tonus sympathique qui majore la thermogenèse du tissu adipeux brun et le brunissement du tissu adipeux blanc(6). Par ailleurs les agonistes sont associés à une moindre accumulation des lipides hépatiques et à une amélioration du métabolisme lipidique systémique(16). Toutefois cette approche entachée par l’effet hyperglycémiant du glucagon n’apparaît pas une bonne approche du traitement de l’obésité. D’où l’intérêt des co-agonistes. Comme l’activation du récepteur du glucagon augmente la dépense énergétique et celle du récepteur du GLP-1 réduit l’apport énergétique tout en neutralisant l’effet hyperglycémiant du glucagon, les agonistes doubles des récepteurs du glucagon et du GLP-1, pourraient être plus efficaces pour traiter l’obésité que les AR-GLP-1 ou que les agonistes doubles des récepteurs GLP-1/GIP comme le tirzépatide qui ont pourtant fait leurs preuves dans le traitement de l’obésité(17). Plusieurs agonistes des récepteurs du GLP-1 et du glucagon sont en phase 1 et 2 de développement, notamment le BI-456906, le pemvidutide, le cotadutide, le SAR425899 et le mazdutide. À titre d’exemple, les essais en phase 2 du survodutide (BI 456906), agoniste double des récepteurs du glucagon et du GLP-1 semblent prometteurs dans le traitement de l’obésité et de la NASH, mais on ne dispose pas encore d’études de non- infériorité par rapport au liraglutide, au sémaglutide ou au tirzépatide(18).

